HFE principles
Human Factors Engineering discovers where foreseeable use errors will occur and implements safety features that prevent normal use errors to result in harm to the user or the patient.
HFE principles, it's not rocket science!
Human Factors Engineering, unlike User experience design, is a use related risk mitigating process and the focus, purpose and added value of of the Human Factors Engineer is clear: minimize use-related hazards and risks and then confirm that these efforts were successful. Scroll down for some information on the history of HFE or go to the introduction page to find some inspiration why you should consider HF engineering as a valuable tool to improve your product and processes.
There are multiple guidelines and standards on applying Human Factors Engineering for the development of your medical device (Regulatory Framework) and they may appear to represent a daunting workload but, in essence, it all can be brought back to a few simple rules and assumptions. It really is not rocket science and, if done right, represents a huge added value to you, rather than a burden.
The FDA summarizes what the focus of Human Factors Engineering should be quite neatly in a simple image which can be translated into: “Consider your User Interface in the hands of the User and the Use Environment, and determine where they might make mistakes that could lead to harm to the patient”.
The fact is that Use errors cannot be avoided and ,for those that could result in harm to the patient, regulators expect manufacturers to identify the potential for use errors and eliminate them by design or mitigate them as much as possible.
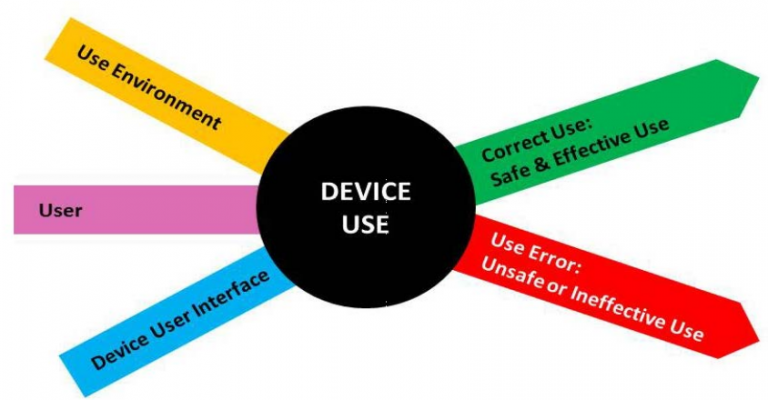
FDA: Applying Human Factors and Usability Engineering to Medical Devices – February 3, 2016
The Weaver approach to HFE
Do it right, but keep it simple and pragmatic.
There are, in my opinion, 2 basic rules-of-conduct to be efficient and successful with the design of your medical device:
1] Sooner is Better!
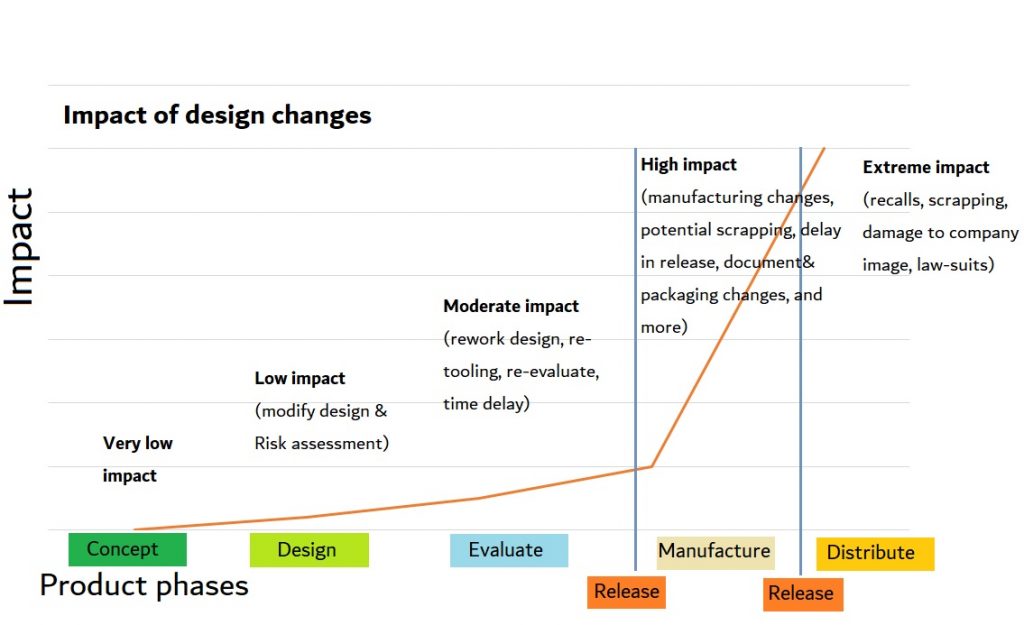
Start as early as possible with analyzing your User Interface, and interacting with users early on to make sure you identify the BIG OOPSES early. Anything you discover in the concept or early design phases can easily and efficiently be translated into value adding, realistic and make-able requirements. Late, mandatory risk mitigating design changes cost a lot of time and money, and headaches.
The ‘traditional’, paper-based User Interface risk assessment can predict many known and expected use problems and is a great way to start focusing on certain design options. But you cannot predict what you don’t know so….
2] Expect the unexpected!
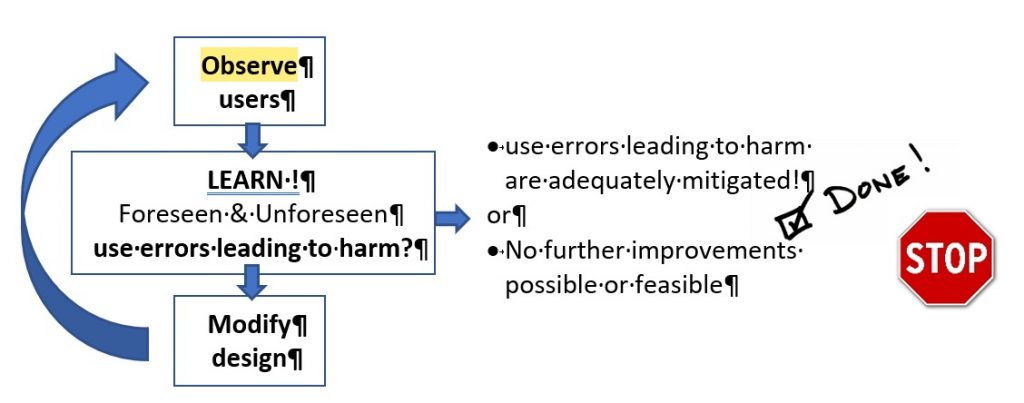
Unanticipated use problems can only, per definition, be found by Interview or observing users when they are confronted with your product (concept idea, wireframe, prototype, development version). The guidelines refer to this as formative usability evaluation testing. So, as soon, and as often as reasonable and useful:
- Get hold of a couple of ‘real’ users
- Ask them to perform representative tasks with a (early) prototype, a sub-part of your device, a single new software screen or a more mature design.
- Observe what the users do, where they succeed, and where they have difficulties with the user interface. Be quiet and let the users do the talking.
- Take the lessons learned and feed them back into your design&development and risk management processes and improve the design to better mitigate use errors that could lead to patient harm.
- Go back to the users and verify whether your design solutions mitigate the use errors.
- Stop when you are confident that the potential for use errors resulting in harm is sufficiently mitigated or no further mitigations are possible.
Follow the process
As mentioned earlier, Human Factors Engineering basically is a use related risk mitigating process with the focus on eliminating or minimizing use-related hazards and risks and then confirm that these efforts were successful.
The guidelines and standards on applying Human Factors Engineering for medical devices (see Regulatory framework), identify and describe the process that you should follow to ensure that you capture these risk, identify risk mitigating measures and evaluate that those risk measures are successful in mitigating the use error from occurring. The guidelines actually make a lot of sense and they help manufacturers structure their efforts and embed HFE thinking into their design&development and risk management processes.
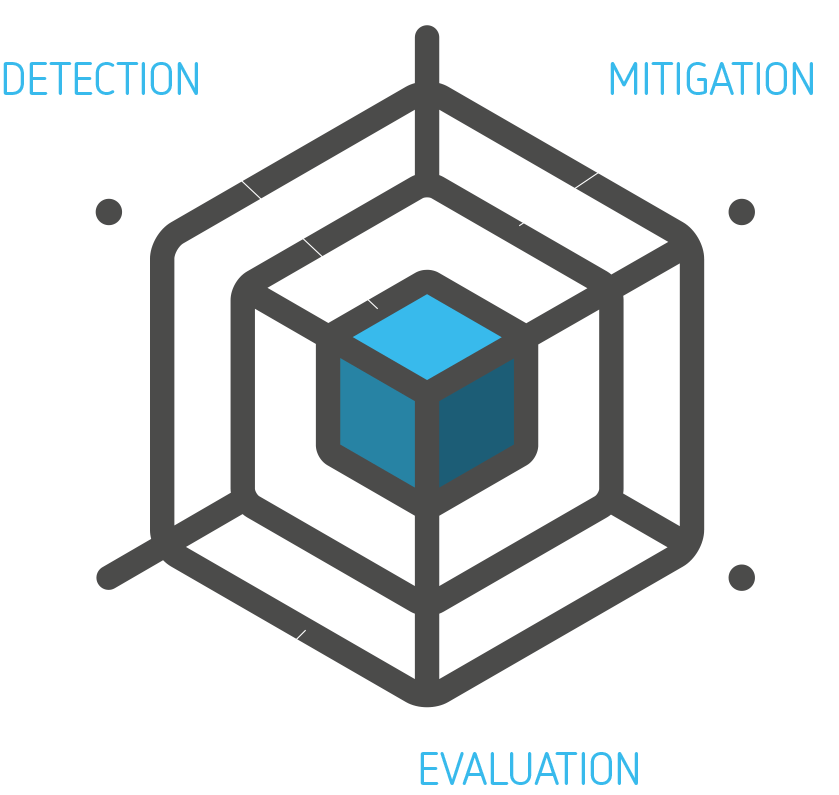
If you follow the process and the 2 rules-of-conduct, you will have much better success at DETECTING and MITIGATING use related risks and EVALUATE that your design choices are actually effective. And when you’re done, ready for launch, the final User Interface EVALUATION (‘summative test’) will provide the prove that you’re design efforts resulted in a product that can be safely allowed onto the market.
I call that the three layers of defense and you can find some details here.